Start-ups und etablierte Pharmaunternehmen sind sich der Bedeutung von Patenten und behördlichen Genehmigungen für ihre Arzneimittelentwicklungsprogramme sehr bewusst. Patente schützen F&E-Investitionen, schließen den Wettbewerb aus und mindern das Risiko. Für die Vermarktung von Arzneimitteln ist eine behördliche Zulassung erforderlich. In den Vereinigten Staaten können sowohl Patente als auch FDA-Produktzulassungen Exklusivrechte für neue pharmazeutische Produkte gewähren.
Die Produktzulassungen der FDA gewährleisten, dass die vermarkteten Arzneimittel sicher und wirksam sind und einem ungedeckten Patientenbedarf entsprechen. Die New Drug Applications (NDA) und die Biologics License Application (BLA) sind die beiden Einreichungswege, die den Sponsoren für die Zulassung ihrer neuen Arzneimittel zur Verfügung stehen. Die beiden Verfahren unterscheiden sich hinsichtlich der zulässigen Produktkategorien, der Zulassungskriterien und der damit verbundenen Vorschriften. Die Marktzulassung für niedermolekulare Arzneimittel fällt unter NDA, während alle biologischen Produkte über das BLA-Antragsverfahren eingereicht werden müssen.
Das Center for Drug Evaluation and Research (CDER), die Abteilung der FDA, die für NDA- und BLA-Anträge mitverantwortlich ist, genehmigt Arzneimittel wie kleine Moleküle und therapeutische Antikörper, nachdem ihre Sicherheit und Wirksamkeit bewertet wurde. Einige dieser Produkte werden als neuartig eingestuft, da es sich um neue molekulare Einheiten (NMEs) und neue therapeutische biologische Produkte handelt. Neuartige Produkte haben Wirkstoffe, die noch nie in den Vereinigten Staaten vermarktet wurden. Andere Originalzulassungen erfolgen in Form von neuen oder erweiterten Anwendungen, Darreichungsformen, Formulierungen und Biosimilars.
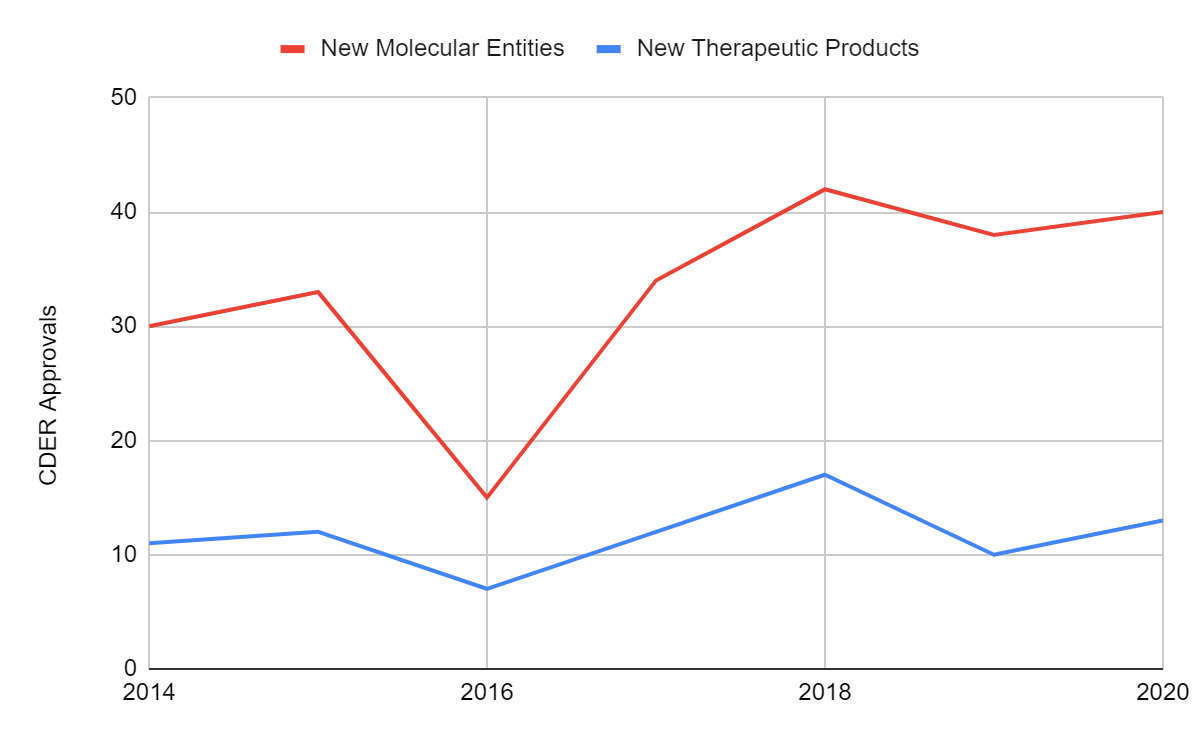
Das obige Diagramm zeigt die neuen Zulassungen für neue Arzneimittel und neue biologische Therapeutika durch das CDER für den Zeitraum 2014-2020. Trotz der COVID-19-Herausforderungen hat das CDER im Jahr 2020 eine Rekordzahl neuer Arzneimittel zugelassen. Vierzig NMEs wurden im Rahmen von NDAs zugelassen, die restlichen 13 waren neue biologische Therapeutika, die im Rahmen von BLAs genehmigt wurden. Die 53 Zulassungen insgesamt liegen nur 6 unter der Rekordzahl von 59 Zulassungen der FDA im Jahr 2018. Der Aufwärtstrend bei den Zulassungen neuartiger Arzneimittel wird sich wahrscheinlich bis 2021 fortsetzen. Zwischen dem 01. Januar 2021 und dem 21. Mai 2021 wurden dreiunddreißig neuartige Arzneimittel und vier neue biologische Therapeutika zugelassen.
Im Folgenden werden einige der jüngsten BLA-Inhaber und die damit verbundenen Patente und Patentanmeldungen vorgestellt.
GlaxoSmithKline
Jemperli von GlaxoSmithKline (GSK) ist ein monoklonaler Antikörper (Dostarlimab-gxly), der den programmierten Todesrezeptor-1 (PD-1) blockiert. Er wurde am 22. April 2021 im Rahmen des beschleunigten Verfahrens zugelassen. Die Behandlung dient der Behandlung von Patientinnen mit rezidivierendem oder fortgeschrittenem Endometriumkarzinom mit Mismatch-Reparaturdefizit (dMMR), bei denen die Behandlung mit einer platinhaltigen Therapie fortgeschritten ist oder die bereits eine solche erhalten haben. Der Antikörper wurde ursprünglich von AnaptysBio entwickelt und später an Tesaro lizenziert, das 2019 von GSK für 5,1 Mrd. USD übernommen wurde.
Dostarlimab wird mit dem in dieser Kategorie führenden Medikament Pembrolizumab (KEYTRUDA; Merck) konkurrieren. Pemrbrolizumab ist derzeit für die Behandlung von Endometriumkarzinomen in Kombination mit Lenvatinib (LENVIMA; Eisai) zugelassen. GSK wird Dostarlimab auch in anderen Indikationen, wie z. B. soliden Krebserkrankungen, einsetzen.
US9815897B2 mit dem Titel "Antibodies Directed Against Programmed Death-1 (PD-1)" (Antikörper, die gegen den programmierten Tod-1 (PD-1) gerichtet sind), das AnaptysBio zugewiesen wurde, beschreibt einen Antikörper, der an den programmierten Tod-1 (PD-1) bindet, verwandte Vektoren und Zusammensetzungen sowie die Behandlung von Krebs oder einer Infektionskrankheit.
ADC-Therapeutika
ADC Therapeutics konzentriert sich auf Antikörper-Wirkstoff-Konjugate (ADC), Behandlungen, die die Selektivität von Antikörpern mit der Wirksamkeit von niedermolekularen Medikamenten kombinieren. Das Unternehmen erhielt seine erste FDA-Zulassung am 23. April 2021. Zynlonta (Loncastuximab Tesirin) wurde im Rahmen eines beschleunigten Verfahrens für die Behandlung von Patienten mit rezidiviertem oder refraktärem (r/r) diffusem großzelligem B-Zell-Lymphom (DLBCL) zugelassen. Bei dem ADC handelt es sich um einen humanisierten monoklonalen Antikörper, der an menschliches CD19 bindet und über einen Linker, eine kurze Peptidsequenz, die zur Bindung des krebsbekämpfenden PBD-Moleküls an den Antikörper dient, mit einem zytotoxischen Sprengkopf auf Pyrrolobenzodiazepinbasis (PBD) konjugiert ist.
Die Behandlung mit einem einzigen Wirkstoff bietet eine Alternative zu bestehenden CD-19-gerichteten CAR-T-Therapien bei DLBCL. Mit dieser Zulassung reiht sich das Unternehmen in eine kleine Gruppe von Unternehmen wie Pfizer, Roche/Genentech und AstraZeneca mit zugelassenen ADC-Produkten ein.
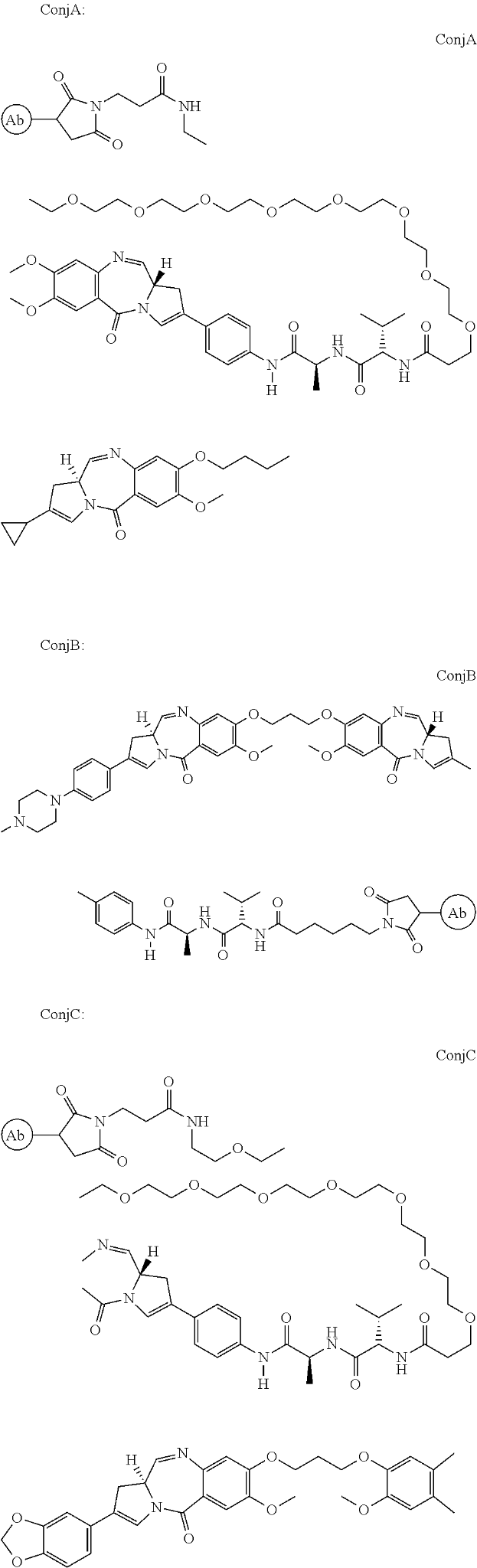
Das Patent US9931414B2 von ADC Therapeutics mit dem Titel "Pyrrolobenzodiazepin-Antikörper-Konjugate" beschreibt Konjugate von PBD, die an CD19-Antikörper binden. Pyrrolobenzodiazepine (PBDs) haben die Fähigkeit, spezifische DNA-Sequenzen zu erkennen und an diese zu binden. Die Anti-CD19-Bindung ist geeignet, das PBD an einen bevorzugten Ort zu bringen, z. B. an einen Krebs, der den CD19-Rezeptor überexprimiert. Das Konjugat ermöglicht die Freisetzung einer aktiven PBD-Verbindung, die keinen Teil des Linkers zurückbehält. Das ADC kann dem Patienten in Kombination mit einer bestehenden Therapie wie Rituximab verabreicht werden.
Regeneron Pharmazeutika
EVKEEZA (evinacumab-dgnb) von Regeneron ist ein injizierbares, verschreibungspflichtiges Arzneimittel, das als Ergänzung zu anderen Arzneimitteln zur Senkung von Lipoproteinen niedriger Dichte (LDL) bei Menschen über 12 Jahren mit einer seltenen Art von hohem Cholesterinspiegel, der sogenannten homozygoten familiären Hypercholesterinämie (HoFH), eingesetzt wird. Bei der familiären Hypercholestroämie (FH) handelt es sich um eine vererbte genetische Erkrankung, bei der der Cholesterinspiegel des Patienten von Geburt an höher als normal ist, da die Leber nicht in der Lage ist, überschüssiges Cholesterin, insbesondere LDL-C, abzubauen oder zu entfernen. Dies hat zur Folge, dass FH-Patienten bereits in relativ jungen Jahren anfällig für Herzerkrankungen sind.
Das Medikament wurde am 11. Februar 2021 von der FDA als bahnbrechend zugelassen. Das Medikament ist auch unter dem Orphan-Status für die Behandlung einer seltenen Krankheit oder eines seltenen Leidens eingestuft. Es handelt sich um einen vollständig menschlichen monoklonalen Antikörper, der an das Angioprotein-ähnliche Protein 3 (ANGPTL3) bindet und dessen Funktion blockiert, wodurch der LDL-C-Spiegel gesenkt wird. Die neue Klasse von Biologika wird mit anderen lipidsenkenden Therapien konkurrieren, wie z. B. den PCSK9-Inhibitorkandidaten: Praulent (Alirocumab) von Regeneron/Sanofi, Repatha (Evolocumab) von Amgen und das zur Zulassung anstehende Leqvio (Inclisiran) von Novartis. Regeneron untersucht das Medikament auch bei refraktärer Hypercholesterinämie und schwerer Hypertriglyceridämie.
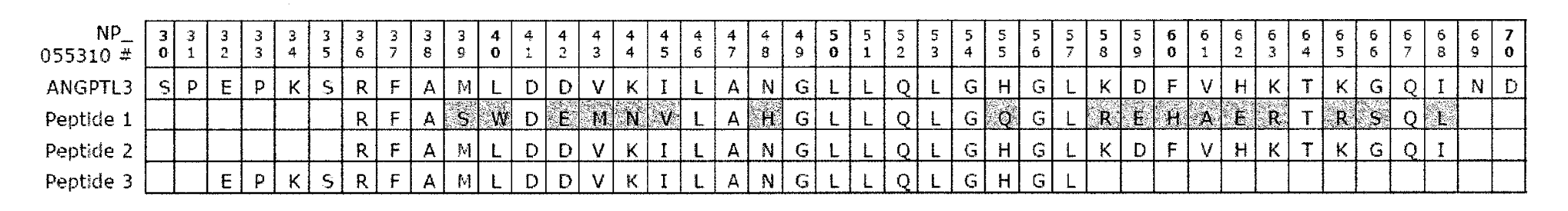
US9018356B2 mit dem Titel "Anti-ANGPTL3-Antikörper und ihre Verwendung" und US20200061189A1 mit dem Titel "Methoden zur Behandlung von Patienten mit familiärer Hypercholesterinämie" beschreiben jeweils einen vollständig humanen Antikörper, der gegen das humane angiopoietinähnliche Protein 3 (hANGPTL3) wirkt, um Krankheiten oder Störungen zu behandeln, wie Hyperlipidämie, Hyperlipoproteinämie und Dyslipidämie, Hypertriglyceridämie, Hypercholesterinämie, Chylomikronämie, Atherosklerose usw.
Johnson & Johnson
Janssen (eine Tochtergesellschaft von J&J) erhielt am 21. Mai 2021 die Zulassung für Rybrevant (amivantamab-vmjw). Es handelt sich um einen vollständig humanen bispezifischen Antikörper, der für die Behandlung von Erwachsenen mit lokal fortgeschrittenem oder metastasiertem nicht-kleinzelligem Lungenkrebs (NSCLC) mit Exon-20-Insertionsmutationen des epidermalen Wachstumsfaktorrezeptors (EGFR) nach einer Platin-Chemotherapie angezeigt ist.
Er konzentriert sich auf eine schwer zu behandelnde Untergruppe von NSCLC-Patienten, die durch den begleitenden, von der FDA zugelassenen diagnostischen Begleittest aus dem Guardant360 CDx Flüssigbiopsie-Bluttest von Guardant Health ermittelt wurde. Der Antikörper zielt auf eine Kombination von EGFR- und Hepatozyten-Wachstumsfaktor-Rezeptoren (MET) ab, während bestehende Tyrosinkinase-Inhibitoren die aberrante EGFR-Signalübertragung von innen heraus angreifen. Die bahnbrechende zielgerichtete Therapie verschafft J&J Zugang zu einem Nischenmarkt von 2-3 % der NSCLC-Patienten. J&J wird sich auch um eine Ausweitung des Einsatzes in anderen Indikationen bemühen, z. B. bei anderen EFGR-mutierten Krebsarten.

US9580508B2 mit dem Titel "Bispecific EGFR/c-Met Antibodies" und US9695228B2 mit dem Titel "EGFR and c-Met Fibronectin Type III Domain Binding Molecules" beschreiben einen isolierten bispezifischen epidermalen Wachstumsfaktor-Rezeptor (EGFR)/Hepatozyten-Wachstumsfaktor-Rezeptor (c-Met) Antikörper, wie z.B. eine bispezifische Fibronektin Typ III (FN3) Domäne, die spezifisch den epidermalen Wachstumsfaktor-Rezeptor (EGFR) und den Hepatozyten-Wachstumsfaktor-Rezeptor (c-Met) bindet und die Bindung von Liganden an die Rezeptoren blockiert. Die Antikörper werden zur Behandlung von Krebserkrankungen wie dem nicht-kleinzelligen Lungenkrebs (NSCLC) eingesetzt. Mutationen können sich in jedem Teil eines EGFR-Gens oder einer mit einem EGFR-Gen assoziierten regulatorischen Region befinden und umfassen Mutationen in den Exons 18, 19, 20 oder 21 oder Mutationen in der Kinasedomäne.
Obwohl Patentinformationen vom NDA-Inhaber als Teil seines FDA-Antrags eingereicht werden, gibt es eine solche Anforderung derzeit nicht für BLA-Inhaber. Selbst wenn solche Informationen verfügbar sind, geben sie keinen Aufschluss über die nicht aufgelisteten Patente/Anmeldungen des NDA/BLA-Inhabers und die therapeutische Landschaft. Eine umfassende Patentrecherche kann das gesamte Ausmaß des Verletzungsrisikos und der Aktivitäten der Wettbewerber aufdecken.
Erfahren Sie mehr über unsere Suchdienste.